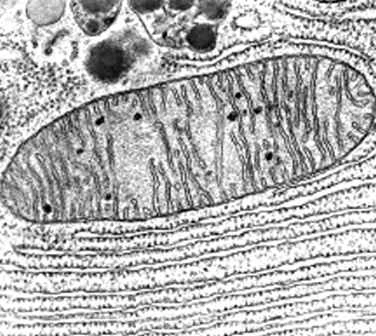
Aparato
de Golgi
El aparato de Golgi es un
orgánulo presente en todas las células eucariotas, el cual pertenece al sistema
de endomembranas y fue visto por primera vez por La Vallete Saint George en
1865 al estudiar los espermatocitos, sin embargo fue Camilo Golgi quien logro
describirlo en 1889 como un “aparato reticular interno”.
La técnica de Schiff-acido
Peryodico marca a los glúcidos de Golgi y esto hace que se puedan ver en el
microscopio óptico.
Está formado por un conjunto
de pilas de sacos aplanados denominados cisternas, éstas varían dependiendo del
tipo celular.
Se divide en dos caras funcionales CIS y TRANS.
Está formado por lamelas,
vesículas, vacuolas, y dictiosomas.
Se divide en dos caras funcionales CIS y TRANS:
Cara
cis
(generalmente convexa) está orientada hacia el retículo endoplásmico y recibe
las proteínas de exportación.
Cara
trans está orientada hacia los gránulos secretorios o los
centriolos
Cisternas
Entre ambas regiones cis y
trans existen cisternas centrales o medias que pueden variar en número.
La cisterna más cercana al
retículo endoplásmico es generalmente fenestrada y presenta continuidad con la
red cis-Golgi (RCG).
En la región trans, el AG se
extiende y forma una red de estructuras tubovesiculares conocida como red
trans-Golgi (RTG).
Las cisternas cis, media y
trans representan una serie de subcompartimentos enriquecidos con enzimas
específicas que llevan a cabo modificaciones postraduccionales a proteínas
recién sintetizadas.
Una de sus funciones es la Glucosilacion.
La función de este organelo está
relacionada con la adición de moléculas de azúcar a los péptidos en tránsito
para la formación de glicoproteínas.
Modelo
de maduración de cisternas
Este modelo habla de que las
cisternas formaban la cara cis de la pila mediante la fusión de los portadores
membranosos desde el retículo endoplasmico
y el ERGIC y que cada cisterna se movía físicamente desde el extremo cis
al trans de la pila y cambiaba de composición conforme avanzaba, por lo tanto
supone que cada cisterna madura a lo largo de la pila.
Modelo
de transporte vesicular
Habla de que el cargamento (proteínas
secretoras, lisosomicas y de membrana) se lanza a través de la pila de Golgi,
desde la CGN hasta la TGN, en vesículas que se desprenden de un compartimento
de membrana y se fusionan con el compartimento contiguo más avanzado de la
pila.
Glucosilacion
La glucosilacion es la
modificación de los hidratos de carbono unidos a glucoproteínas y glucolipidos
sintetizados en el retículo endoplásmico.
Glucosiltransferasas: incorporan residuos glucídicos específicos
Glucosidasas: eliminan residuos glucídicos específicos.
La secuencia en que se
transfieren los azucares durante el ensamblaje de un oligosacárido depende de
la secuencia de acción de las glucosiltransferasas que participan en este
proceso.
Hay dos
familias: N-glicoproteínas y O-glicoproteínas
Depeniendo del lugar de
adición de los carbohidratos:
N-glicoproteína: los carbohidratos se unen al grupo
amino de la cadena lateral del aminoácido asparagina.
O-glicoproteína: en este caso, el punto de unión es
el grupo hidroxilo de las cadenas laterales de los
aminoácidos serina y treonina.
A diferencia de los
oligosacaridos con enlaces N, cuya síntesis comienza en el RE, los unidos con
proteínas mediante enlaces O se articulan por completo dentro del aparato de
Golgi.
Primer
paso:
Después del retiro de los
tres residuos de glucosa,
Glucosidasa 1 quita un
residuo de glucosa en el retículo endoplasmatico
Glucosidasa 2 quita dos
residuos de glucosa.
Manosidasa quita una manosa
del oligosacárido formado
Glucosilación
en aparato de Golgi
Primer
paso
El oligosacarido pasa a la
red Cis Golgi
Enzima Manosidasa actúa quitando varios residuos de manosa en la región
CIS Golgi.
segundo
paso
Enzima N-acetil-glucosamina transferasa I actúa agregando
un residuo de N-acetilglucosamina a uno de los residuos de manosa en la región
CIS Golgi.
Tercer
paso
Interviene la enzima
Manosidasa II retirando dos residuos de manosa en el oligosacárido en la región
medial Golgi.
Cuarto
paso
Actúa la enzima
N-acetil-glucosamina transferasa II agregando otro residuo de N-acetilglucosamina a la manosa
que quedo libre en la región medial
Trans Golgi.
Quino
paso
La enzima fucosil -y
-galactosil -transferasa en la región Trans Golgi agrega residuos de galactosa
para obtener un oligosacárido complejo.
Sexto
paso
Enzima Sialil-transferasa
agrega un ácido sialico en la región más Trans de Golgi.
Transporte
vesicular en el aparato de Golgi
Mediado por pequeñas
vesículas de gemación desde un compartimento de membrana (donador) el cual
produce las vesículas para fusionarse con un compartimento (aceptor) que recibe
la vesícula y su contenido.
Los materiales son transportados
entre compartimentos por vesículas que se desprenden de membranas donadoras y
se fusionan con las membranas receptoras.
Gemacion
de vesículas
Forma parte fundamente en el
transporte vesicular, y para que se lleve a cabo la formación una vesícula, se
requiere de la participación de proteínas de recubrimiento. Ésta
cubierta es utilizada como medio mecánico para que las vesículas puedan dirigirse a su destino.
Función
de cubiertas de proteínas
Sirven
como dispositivo mecánico que hace que la membrana se curve y forme una
vesícula desprendible.
Proporciona
un mecanismo para seleccionar los componentes que transporta la vesícula.
Incluye
como cargamento en proteínas secretoras, lisosomicas y de membrana que
deben transportarse.
La
cubierta de esta vesícula se encuentra formada por dos capas distintas de
proteínas: una jaula externa que forma el marco de la cubierta y una capa
interna de adaptadores que sirven para unir la cara externa de la bicapa lipídica
y el cargamento de vesículas.
Vesículas
cubiertas con COP I
Mueven materiales en sentido
retrogrado: del ERGIC y la pila de Golgi “hacia atrás” al ER y de las cisternas
Golgi trans “de regreso” a las cisternas Golgi cis.
Fueron identificadas por
primera vez en experimentos en los que las células se trataron con moléculas de
estructura similar al GTP pero a diferencia de este, no pueden hidrolizarse.
Vesículas
cubiertas con clatrina
Movilizan materiales de la
TGN a los endosomas,lisosomas y vacuolas vegetales.
También mueven materiales de
la membrana plasmática a los compartimentos citoplasmáticos a lo largo de la
vía endocitica.
Además se han implicado en
el tránsito de los endosomas y lisosomas
Vesículas
cubiertas con COP II
Desplazan materiales del retículo
endoplásmico “hacia adelante¨al ERGIC y al aparato de Golgi.
Conservación
y recuperación de las proteínas residentes del retículo endoplasmico
Se sugiere que influyen dos
mecanismos:
Retención
de moléculas residentes que se excluyen de las vesículas de transporte.
Recuperación
de moléculas prófugas
Las proteínas solubles
residentes de la luz del ER como la disulfuro isomerasa de proteínas casi
siempre tienen la señal de recuperación KDEL (lys-asp-glu-leu).
Ordenamiento
de proteínas en la red trans de Golgi (TGN)
Esta red es la última
estación del aparato de Golgi, funciona como una instancia clasificadora y
dirige las proteínas hacia diversos destinos.
Un ejemplo es la vía de dirección
de las enzimas lisosómicas a los lisosomas
Paso
1
Los residuos de manosa de la enzima lisosomica
se fosforilan en las cisternas de Golgi.
Paso
2
Los residuos de manosa luego
se incorporan en forma selectiva en la vesícula cubierta con clatrina en la
TGN.
Paso
3
Se cree que los receptores
para la manosa 6-fosfato tienen doble función.
Paso
4
Los receptores de la manosa
6-fosfato interactúan de manera específica con las enzimas lisosomicas en el
lado luminal de la vesícula y con los adaptadores en la superficie citosólica
de la vesícula. Los receptores de la manosa 6-fosfato se separan de las
enzimas.
Paso
5
Una vez que se separan los
receptores para manosa-6 fosfato de las enzimas estos regresan al aparato de
Golgi.
Paso
6
Las enzimas lisosomicas se
vacían en un endosoma y al final en un lisosoma.
Paso
7
Los receptores para manosa
6-fosfato también están presentes en la membrana plasmática donde pueden
capturar enzimas lisosomicas que se secretan hacia el espacio extracelular y
regresan las enzimas a una vía que las dirige a un lisosoma.
Vesículas
cubiertas con Clatrina
Contienen:
Una celosía externa parecida
a un panal formada por la proteína clatrina, la cual constituye un soporte
estructural.
Una capa interna formada por
adaptadores de proteína que cubre la superficie de la membrana de la vesícula y
que está dirigida hacia el citosol.
Vesícula
con cubiertas COP II
Selecciona y concentra
ciertos componentes para transportar en vesículas.
Las vesículas cubiertas con
COPII median la primera rama del traslado por la vía biosintética, del RE al
ERGIC y la red cis de Golgi.
Las proteínas seleccionadas
por estas vesículas incluyen:
-Enzimas que actúan en las
etapas avanzadas de la vía biosintética, como las glucosiltransferasas del
aparato de Golgi.
-Proteínas de membrana que
pueden unirse con cargamento soluble.
Ejemplo: Proteína G llamada
Sar1, que tiene función reguladora, en el inicio de la formación de la vesícula
y la regulación del ensamblaje de la cubierta de la vesícula.
Formación
de vesículas
1.- Sar1 es reclutada en la
membrana del RE en la forma unida a GDP y es inducida a intercambiar su GDP por
un GTP por una proteína llamada GEF (factor de intercambio de guanina) ( al
unirse al GTP, Sar1 sufre un cambio que hace que la hélice alfa en su extremo N
terminal se inserte en la hoja citosolica de la bicapa del RE.
2.- Se induce la curvatura
de la bicapa lipidica en ese sitio. (Formacion de una membrana aplanada a una
esferica).
3.- Sar1-GTP atrae dos
polipeptidos adicionales a la cubierta COP II, Sec23 y Sec24, que se unen con
forma de banana, ( Sec24 funciona como proteína adaptadora de la cubierta COPII).
4.- Las subunidades
restantes de la cubierta COPII Sec23 y Sec 31, se unen con la membrana para
formar la jaula estructural externa de la cubierta proteínica.
Después de esto, se ensambla
toda la cubierta COPII la yema se separa de la membrana del RE en la forma de
una vesícula cubierta por COP II.
Enfermedades
relacionadas
Coroideremia
Se
caracteriza por presentar invariablemente degeneración retiniana y de coroides. Determinada
genéticamente, con un modo de herencia recesivo ligado al cromosoma X.
El primer signo de
afectación ocular consiste en ceguera nocturna que inicia en la infancia
temprana que progresivamente conduce a la ceguera total alrededor de los 20 o
incluso hasta los 45 años.
Síndrome
de Lowe (síndrome oculocerebrorrenal)
Trastorno genético heredado
en forma recesiva ligada al cromosoma X.
Entre sus complicaciones se
encuentran: cataratas congénitas,
disfunción renal tubular y déficit neurológico, proteinuria, aminoaciduria,
fosfaturia y acidosis metabólica, así como retardo mental, hipotonía,
trastornos de la conducta y ausencia de reflejos osteotendinosos profundos.
Enfermedad
de Menke
Se caracteriza por la
incapacidad de las células para liberar el cobre absorbido, debido a un defecto
en el gen ATP7A.
El
cobre se puede acumular en el intestino delgado y los riñones, pero los niveles
bajos de este elemento en otras zonas pueden afectar la estructura de huesos,
piel, cabello y vasos sanguíneos e interferir con la función nerviosa.
Mulcolipidosis
II
Se caracteriza por la
presencia de múltiples inclusiones en el citoplasma de los fibroblastos. Se debe a que existe una señalización
anómala de enzimas lisosomales en las células del tejido mesenquimatoso, como
las hidrolasas lisosomales durante su paso por Cis-golgi adquieren residuos de
manosa 6-fosfato.
Mulcolipidosis
III
Se presenta a muy temprana
edad
Retraso
mental progresivo
Malformaciones
esqueléticas
Los pacientes mueren en la
primera década de vida.
Anomalías cardiovasculares
Presenta una progresión más
lenta en el deterioro mental, viven hasta la edad adulta.
Alteraciones en las glicoproteínas
séricas (exportación) que consisten en
un peso molecular menor al esperado debido a un bajo nivel de glucosilacion.
Complicaciones de la
enfermedad:
Retraso
psicomotor
Alteraciones
dermatológicas
Oftalmológicas
Disfunción hepática

Bibliografía
Gerald Karp, Biología celular y molecular, MC Graw Hill Education, México, 2014, pag: 290-300