Mitocondria
El origen de las
mitocondrias está apoyado en la teoría de que una
célula procariota se fusionó en un momento de
la evolución con otra célula procariota o eucariota.
Sus fundamentos están
basados en que las bacterias y las mitocondrias tienen mucho en común, tales
como el tamaño, la estructura, componentes de su membrana y la forma en que
producen energía, etc.
Las mitocondrias poseen su
propio ADN y están recubiertas por su propia membrana.
El código genético del ADN
mitocondrial suele ser diferente al
código genético del ADN nuclear.
Función
Captación y liberación de
iones de calcio
Síntesis de sustancias
Generación de moléculas de ATP
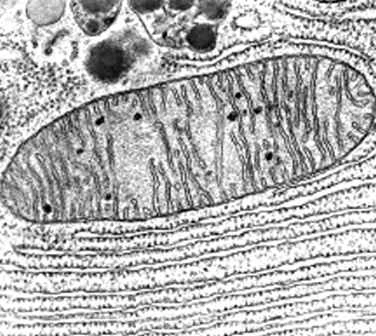
Está
conformada por:
Una membrana externa
Espacio intermembrana
Una membrana interna
Crestas
Matriz mitocondrial
Membrana mitocondrial
interna se encuentra subdividida en: membrana limitante interna y crestas
Membrana limitante interna: importa proteínas mitocondriales
Crestas: ayudan
en la respiración aeróbica, formación de ATP y ayudan a incrementar las áreas
de la superficie.
Espacio intermembrana: tiene la misma composición que el citoplasma de una célula, y sus proteínas son conocidas por su participación en la muerte celular.
Matriz
mitocondrial
Contiene: ribosomas, moléculas
de DNA (las mitocondrias tienen su propio material genético) y codifican 24 RNA.
Transporte
de lípidos y proteínas en la mitocondria
El piruvato y los ácidos
grasos se transportan desde el citosol a las mitocondrias a través de la membrana
mitocondria externa.
Se transforman en acetil
coenzima A (acetil CoA) en la matriz mitocondrial por acción de las enzimas del
ciclo del ácido cítrico. De modo que la célula libera CO2 como desecho
metabólico.
La translocasa de la
membrana mitocondrial externa (TOM) constituye la vía habitual de entrada de
proteínas precursoras codificadas por genes del núcleo celular.
Captación
de proteínas en las mitocondrias
Existen cuatro
compartimientos a los cuales pueden llegar las proteínas:
OMM
IMM
Espacio intermembranoso
Matriz
Las proteínas mitocondriales
cuentan con una secuencia directriz removible, llamada presecuencia situada en el extremo N de la molécula.
Esta presecuencia comprende
varios residuos con carga positiva en una cara de la hélice y residuos
hidrófobos en la cara contraria.
Importancia
de las coenzimas reducidas en la formación de ATP
En la membrana interna de la
mitocondria las coenzimas reducidas son utilizadas en la Cadena Transportadora
de Electrones y fosforilación oxidativa para la producción de ATP.
Lanzadera de Malato-Asparatato
y Fosfato de glicerol
Cadena Transportadora de
Electrones y Fosforilación Oxidativa
Perspectiva
Humana
La contracción muscular
requiere grandes cantidades de energía, por lo que en la contracción máxima la
velocidad de hidrólisis de ATP aumenta hasta 100 veces.
Metabolismo
Anaeróbico:
Se realiza por medio del
Ciclo de Cori
La formación de ácido
láctico provoca un descenso del pH (7-6.3) del tejido muscular.
Metabolismo
Aeróbico
Se realiza por medio de la oxidación
de la glucosa
Una vez que se degradan las
reservas de glucógeno, continúa con el catabolismo de los ácidos grasos
provenientes del tejido adiposo.
Transporte
de Electrones
Las enzimas deshidrogenasas
transfieres electrones de sustratos a coenzimas
Ciclo de Krebs 5 y reacciones
que se llevan a cabo por hidrolasas
Generan NADH: Isocitrato DH,
α-cetoglutarato DH y Malato DH
Genera FADH2: Succinato DH
Electrones de alta energía
unidos al NADH y FADH2 son transferidos por acarreadores específicos de
electrones que conforman la Cadena Transportadora de Electrones.
Tipos
de Portadores de Electrones
Flavoproteínas
Polipéptido unido con fuerza
a un FAD
Es capaz de aceptar y donar
2 electrones y 2 protones
La deshigrogenasa de NADH y
Succinato DH, son las principales flavoproteínas
Citocromos
Proteínas con grupos
prostéticos hemo
Existen citocromos a, b y c
Tres
átomos de cobre
Pertenecen al complejo
proteínico de la membrana interna
Aceptan y donan un electrón
dependiendo del estado.
Ubiquinona
Molécula liposoluble con una
cadena hidrófoba larga compuesta de unidades isoprenoides de 5 carbonos.
Pueden aceptar y donar 2
protones y 2 electrones
Proteínas
con Hierro y Azufre
Los núcleos llegan a tener
2-4 átomos de hierro y azufre
Es capaz de aceptar y donar
un electrón
Se han identificado >12
centros de hierro-azufre diferentes en la mitocondrias
Complejos Transportadores de
Electrones
Complejo I – Deshidrogenasa
de NADH
Puerta
de entrada a la Cadena Transportadora de Electrones
Transferencia
de un par de electrones del NADH a la Ubiquinona para formar Ubiquinol
Conglomerado
enorme en forma de “L”, contiene 45 subunidades diferentes y presenta una masa
molecular de 1 millón Da.
Los
componentes para la transferencia de los electrones están dentro del segmento
hidrófilo
Los electrones se
transfieren por 7 centros de hierro-azufre
Los electrones se
transfieren a una molécula de ubiquinona
Se acompaña con 4 protones
desde la matriz al espacio intermembranal.
Complejo
ll (Deshidrogenasa de Succinato)
Está
formado por cuatro polipéptidos: dos subunidades hidrófobas
que fijan la proteína a la membrana y dos subunidades hidrófilas que comprenden
la enzima del TCA.
Suministra una vía para
alimentar al FAD y la ubiquinona con electrones de baja energía.
Se mueven los electrones a
través de 3 cúmulos de hierro azufre.
También tiene un grupo hemo,
el cual atrae a los electrones que se han escapado
No se acompaña de
translocación de protones
Complejo
lll (Citocromo bc1)
Transferencia de electrones
del ubiquinol al citocromo c.
Se bombean 4 protones por
cada 2 electrones que pasan por el complejo lll.
Dos protones provienen de la
molécula de ubiquinol que ingreso al complejo y dos se retiran de la matriz
como parte de la segunda molécula de ubiquinol.
Complejo
lV (Oxidasa de citocromo c)
El paso final en el
transporte de electrones en una mitocondria es la transferencia de dichas
partículas del citocromo c reducido al
oxigeno
El complejo IV o citocromo c
oxidasa; capta cuatro electrones de las
cuatro moléculas de citocromo c y se transfieren al oxígeno (O2), para producir
dos moléculas de agua (H2O).
Al mismo tiempo, se translocan cuatro protones
al espacio intermembrana, por los cuatro electrones.
Además
"desaparecen" de la matriz 2 protones que forman parte del H2O.
Translocación
de protones y establecimiento de la fuerza protón-motriz
La
translocación de protones a través de la membrana interna es electrogénica
porque aumenta la cantidad de cargas positivas en el espacio intermembrana y el
citosol e incrementa la cantidad de cargas negativas dentro de la matriz.
Hay dos componentes del
gradiente de protones que deben considerarse
.-La diferencia de
concentraciones de iones de hidrogeno entre un lado de la membrana y el otro.
.-El voltaje, que se produce
por la separación de carga a través de la membrana. Un gradiente tienen un
componente de concentración químico y el otro eléctrico a esto se le denomina
gradiente electroquímico
La energía de ambos
componentes del gradiente se expresa como fuerza protón-matriz que se mide en
mini voltios.
La contribución del
potencial eléctrico a la fuerza protón-matriz comparada con la participación
del gradiente pH depende de la permeabilidad de la membrana interna.
Por ejemplo en el movimiento
de protones hacia el exterior durante el transporte de electrones se acompaña
con carga negativa.
Para que se mantenga una
fuerza protón matriz es necesario que la membrana mitocondrial interna
permanezca impermeable a los protones.
De
lo contrario el gradiente establecido se disipa debido al escape de protones de
regreso a la matriz, lo que conduce la liberación de energía en forma de calor.
Mecanismo
para la formación de ATP
En el año de 1960 Humberto
Fernández Moran descubrió una capa de esferas unidas mediante tallos a la cara
interior de la membrana interna
Efrain Racker: aisló las
esferas de la membrana interna a las que llamo F1 y se comportaban como una
ATPasa.
Si se considera que la
hidrolisis del ATP es la reacción inversa a su formación, la función de las
esferas F1 se torna más evidente, contiene el sitio catalítico en el que ocurre
por lo general la formación de ATP:
1.- Las enzimas no afectan
la constante de equilibrio de la reacción que catalizan
2.- Las enzimas son capaces
de catalizar las reacciones en ambos
sentidos
Estructura
de la sintasa de ATP
ATP (Adenosina Trifosfato)
es la principal molécula de la que obtienen y almacenan energía los seres
vivos. La degradación a ADP y fosfato inorgánico del ATP genera energía que los
enzimas del organismo aprovechan para realizar las funciones que requieren un
aporte energético.
La enzima encargada de
sintetizar esta molécula en las células se denomina ATP sintasa. Normalmente al romper moléculas complejas se libera
energía en forma de protones que la ATP sintasa aprovecha para generar ATP, que
será almacenado para su uso posterior
La ATP sintasa es un complejo
grande, de unos 371 KDa, formada por varias subunidades divididas entre la
unidad bombeadora de protones, H+, llamada F0 que se encuentra en la membrana y
la unidad catalítica F1 en el citoplasma o en el lumen del orgánulo.
- La unidad catalítica, F1, está formada a su vez por 5
tipos de subunidades. Las 3 subunidades alfa y las 3 beta pertenecen a la
familia de las NTPasas (proteínas de formación de bases trifosfatos).
Las subunidades alfa y las
beta son las encargadas de unir el ADP y el fosfato inorgánico, aunque en la
ATP sintasa tan solo las subunidades beta participan en la catálisis del ATP.
Las 3 subunidades beta se generan a partir de 3 genes diferentes, aunque las
cadenas de aminoácidos son equivalentes.
Además la subunidad catalítica cuenta con una
subunidad gamma, una delta y una épsilon. Las subunidades gamma y épsilon
anclan F1 a F0, formando un tallo.
Además la subunidad gamma es
la encargada de la rotación de la unidad sintetizadora, con lo que modifica a
la subunidad beta para que acepte ADP.
Fundamento
de la formación de ATP según el mecanismo de cambio de unión
1.- La energía liberada por
el movimiento de los protones no se usa para impulsar la fosforilacion del
ADP en forma directa, sino que se emplea
para cambiar la afinidad de unión del sitio activo por el producto
2.- Cada sitio activo adquiere de manera sucesiva
tres diferentes conformaciones con afinidades distintas por los sustratos y los
productos.
3.- El ATP se sintetiza por
catálisis rotatoria, en la que una parte de ATP sintasa gira con respecto a
otra parte.
Funciones
de la fuerza protón-motriz además de la síntesis de ATP
1.-Promueve la captación de
ADP y P i en las mitocondrias a cambio de ATP e H+
2.-Puede usarse como fuente
de energía para atraer iones de calcio
3.-Impulsa los eventos que
inducen la función mitocondrial
4.-Hace que polipéptidos
entren a la mitocondria desde la matriz
Reproducción
mitocondrial
Existen pruebas de que las
mitocondrias se reproducen antes de la mitosis para responder al aumento de las
necesidades metabólicas de la célula y para sustituir a mitocondrias viejas,
que terminan siendo degradadas en citolisosomas.
La composición de la
membrana mitocondrial externa es más parecida a la del retículo endoplasmático,
mientras que la interna se parece más a la membrana plasmática de las células
procariotas.
Los componentes de la matriz
mitocondrial son mucho más semejantes a los del citoplasma de las células procariotas
que a los del citoplasma de las células eucariotas.
La cadena transportadora de
electrones y la ATP sintetasa de la membrana interna y crestas mitocondriales
tienen su equivalente en el mesosoma bacteriano.
Las mitocondrias de células
eucariotas muy diferentes entre sí contienen en su membrana proteínas de antígenos
en común.
Importancia
clínica: Herencia mitocondrial
Las mitocondrias se
transfieren a través de la madre. Las enfermedades mitocondriales pueden
afectar tanto a hombres como a mujeres, aunque los primeros no pueden
transmitirlas
Epilepsia mioclónica con fibras rojas rasgadas (MERRF)
La MERRF se debe a una mutación puntual de un
gen del cromosoma mitocondrial que codifica ARNt para lisina. La anomalía en el
ARNt altera la síntesis de las proteínas implicadas en el transporte de electrones
y la producción de ATP.
Neuropatía óptica hereditaria de Leber (LHON)
La enfermedad se restringe
al ojo. Los pacientes afectados presentan una pérdida súbita de visión en la
segunda o la tercera década de la vida.
Síndrome médula-páncreas de Pearson
Anemia y miopatía mitocondrial observadas
durante la infancia.
Leber
Entre los 18 y los 30 años
se observa con frecuencia una pérdida de visión central aguda o subaguda,
brusca e indolora.
Afecta a ambos ojos
simultáneamente o de forma secuencial, con pérdida de visión en el segundo ojo
semanas o meses después.
También pueden darse otros
signos neurológicos.
Estas anomalías se conocen como Leber
"plus", e incluyen trastornos
motores, distonía, temblor postural y ataxia cerebelosa.
Pearson
Disturbio mitocondrial
congénito, de carácter raro, que se da normalmente como consecuencia de un
rearreglo del ADN mitocondrial o también pero menos frecuentemente por causa de
mutaciones. Se caracteriza por una anemia
sideroblástica refractaria, vacuolización de los precursores de la médula ósea
y disfunción exocrina del páncreas.
Barth
Causado por mutaciones en el
gen TAZ (tafazzin; Xq28) que codifica la aciltransferasa Taz1p implicada en el
metabolismo de la cardiolipina, un fosfolípido muy importante de las membranas
mitocondriales internas, por lo tanto compromete a la estructura mitocondrial o
la función de la cadena respiratoria.
Síndrome
de Leigh
Síntomas son la falta de
succión en los bebés y la pérdida de control de la cabeza y de los movimientos
voluntarios.
Pérdida de apetito, vómitos,
irritabilidad, llanto continuo y convulsiones.
Si la enfermedad progresa,
los síntomas incluyen debilidad generalizada, falta de tono muscular,
espasticidad, trastornos del movimiento, incapacidad para coordinar el equilibrio.
Se puede clasificar en:
ADN mitocondrial o Síndrome
de Leigh de transmisión (o herencia) materna
ADN nuclear: causada por una
mutación en el ADN nuclear, y sub-dividido de acuerdo al modo de herencia y del
gen mutado en la forma con herencia autosómica recesiva y la forma con herencia
ligada al cromosoma X.
Síndrome
de Kearns-Sayre
Síntomas: sordera bilateral neurosensorial, las
afectaciones cardiacas, las afectaciones del sistema nervioso central, la
miopatía del músculo esquelético, los desórdenes intestinales y endocrinos y el
fallo renal.
Causado por delaciones de
grandes fragmentos de ADN mitocondrial (ADNmt), resultando en la pérdida de
genes involucrados en la ruta de señalización de la fosforilación oxidativa.
Tratamiento: colocación de
un marcapasos, proporcionar audífonos. El suplemento con coenzima Q10 ha
resultado ser beneficioso en algunos casos. Las manifestaciones oftalmológicas
se pueden tratar quirúrgicamente
Encefalopatía
Mitocondrial
Herencia: La mutación
3271T> C del gen ARNt Leu
Síntomas: cefaleas con vómitos y, en ocasiones, signos de pseudoapoplejía, como
confusión, hemiparesia y hemianopsia.
A menudo se producen en
pacientes con síntomas crónicos como debilidad muscular, sordera, diabetes,
baja estatura, miocardiopatía.
No existe ningún tratamiento
que hasta hoy haya sido exitoso
Bibliografía
Gerald Karp, Biología celular y molecular, MC Graw Hill Education, México, 2014, pag: 178-207




la infertilidad para facilitar la concepción. (No más adopción, con el Dr. Itua su problema se resolverá y tendrá su hijo con facilidad.He sido sitios de blog por un tiempo y hoy me sentí como que debía compartir mi historia porque yo era una víctima también. Tuve endometriosis durante 18 años y nunca pensé que alguna vez tendría una cura debido a los terribles síntomas que tenía y esto hizo imposible que me quedara embarazada incluso después de 12 años de matrimonio y fue un problema grave. Me enteré de que el Dr. Itua en el sitio de blog que trató a alguien y la persona compartió una historia de cómo consiguió una cura y dejó sus datos de contacto, me puse en contacto con el Dr. Itua y él realmente lo confirmó y decidí dar una oportunidad también y utilizar su medicina a base de hierbas que fue como mi carga terminó por completo. Mi hijo cumplirá 2 años este diciembre y además yo sufría de cáncer cerebral, lo cual también le explico al Dr. Itua, así que él me preparó una medicina a base de hierbas que bebí durante dos semanas para curar mi cáncer de Brian, así que si usted sufre de cualquier tipo de enfermedad puede contactar con el centro de hierbas del Dr. Itua para obtener su cura con éxito, estoy agradecida a Dios y agradecida a su medicina también. El Dr. Itua también puede curar las siguientes enfermedades... Cáncer, VIH, Herpes, Epilepsia, Hepatitis B, Inflamación del hígado, Diabetes, Fibroides, Disfunción eréctil, Recuperar a tu ex, si tienes (A sólo contacta con él en (drituaherbalcenter@gmail. com o en el número Whatsapp..+2348149277967)También puede aconsejarte sobre cómo manejar algunos problemas maritales.
ResponderBorrar